Diagnosis
Identifying patients for diagnostic testing remains a challenge in clinical practice, contributing to diagnostic delays and misdiagnoses.14,15
Although patients are commonly diagnosed in childhood or adolescence, the average age at diagnosis for the most common type of GD (type 1) is 30-40 years of age.9
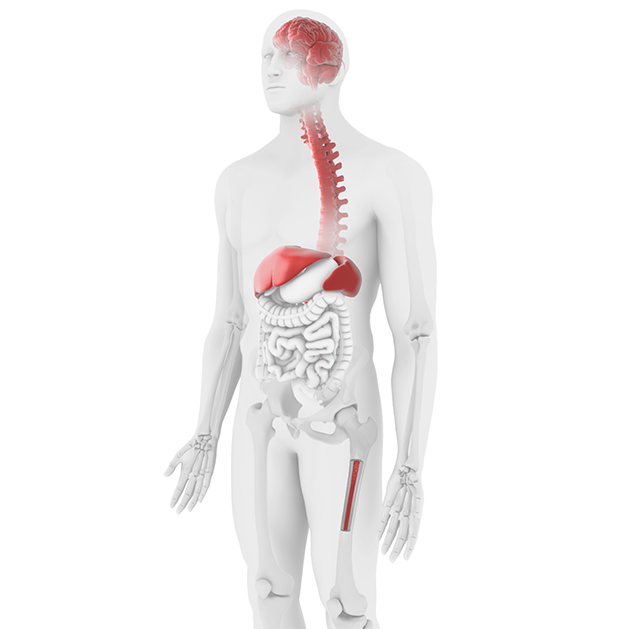
Initial diagnosis is based on laboratory testing and physical examination including:16
- hematologic (complete blood count, liver function test)
- skeletal (bone mineral density, magnetic resonance imaging (MRI) of hip/spine)
- visceral (MRI of spleen/liver)
- cardiopulmonary (electrocardiogram, chest x-ray)
- central nervous system (neurological exams)
However, a definitive diagnosis requires enzymatic analysis of glucocerebrosidase and can also be supplemented by detection of genetic defects.17
Type 2 GD, with substantial CNS involvement, is typically diagnosed in infancy, with bulbar signs and oculomotor paresis, while Type 3 GD is typically diagnosed in childhood or adolescence, often with a progression of CNS involvement.11