
American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM), 2026
Annual meeting dedicated to the advancement of neuromuscular, musculoskeletal, and electrodiagnostic medicine.
To report an adverse event or product quality complaint, call 1-877-TAKEDA-7 (1-877-825-3327)
To report an adverse event or product quality complaint, call 1-877-TAKEDA-7 (1-877-825-3327)

Not actual patients.
Angioedema attacks most commonly affect the face, extremities, abdomen and urogenital area and are often painful and debilitating.1,2
Less commonly, they can occur in the laryngeal area causing airway obstruction and potentially becoming life-threatening.2
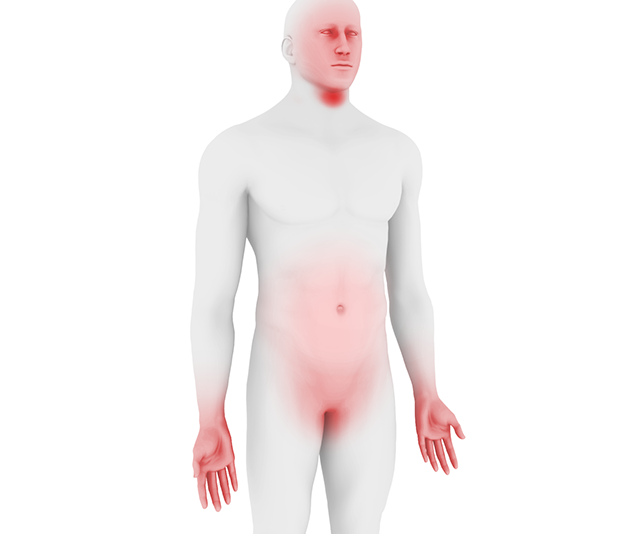

HAE is a rare condition with an estimated prevalence of 1 in 50,000, though exact prevalence may vary due to misdiagnosis and geographical region.1,2,3 Symptoms are unpredictable and can manifest at any age, but typically appear during childhood (5 to 11 years), worsen during puberty (particularly among females), and persist throughout life.2,4,5,6,7

HAE is an autosomal dominant disorder resulting from a deficiency of C1 inhibitor (C1-INH), which is a naturally occurring protein that inhibits plasma kallikrein, a key mediator of inflammation.1,8,9,10,11 C1-INH deficiency results in uncontrolled kallikrein activity, leading to increased generation of the vasoactive peptide bradykinin and associated symptoms of pain and swelling.8,9,10,11,12
The disease is marked by its genetic variability – more than 510 different mutations of the C1-INH gene (SERPING1) have been identified.13 However, it is generally classified into 3 types according to the underlying cause and levels of C1-INH:

Due to its rarity and symptom overlap with other conditions, HAE is frequently under-recognized leading to delayed diagnosis.4,7,16,17 Historically, the rate of mortality has been higher in undiagnosed HAE patients due to lack of awareness of potential fatal laryngeal attacks and inadequate treatment.18 The two most common forms of HAE (types I/II) should be suspected when a patient presents with a history of recurrent angioedema attacks and further substantiated by:19
To confirm diagnosis, patients should be assessed for blood levels of C1-INH protein, C4 and C1-INH function.19

The condition is associated with a substantial and multifaceted burden of illness affecting various aspects of a patient's quality of life (e.g., emotional state, career and education progression, and the decision to have children).2,7,20,21,22,23,24 Types I/II are indistinguishable in their clinical presentation, having identical symptoms characterized by edema attacks that can vary in location, frequency, duration, and severity.1,25,26 Swelling and other symptoms gradually worsen over 12 to 36 hours, intensifying in a relentless manner, sometimes spreading to other sites, and then resolving over 2 to 5 days.7,14,27
A variety of possible triggers for attacks have been proposed, most commonly mechanical trauma, mental stress, and airway infection.2,4,28 Nevertheless, many attacks occur without an obvious trigger, particularly in children, highlighting the need for individualized treatment plans.2,4,19
This resource provides information on Takeda medications available in the Hereditary Angioedema category and is not intended to represent a complete list of therapeutic options.
[C1 Esterase Inhibitor (Human)]
(icatibant)
(ecallantide)
(lanadelumab-flyo)
This is not intended to be a comprehensive resource of all congresses and congress materials across therapeutic and disease areas. Congress materials may include information about investigational use(s) of compounds/products that are not approved for use by the U.S. Food and Drug Administration (FDA) and/or are inconsistent with the Prescribing Information. Takeda does not recommend the use of any Takeda product beyond the approved labeling. Any decisions regarding the usage of a Takeda product beyond the approved labeling are left to the discretion of the healthcare professional. Takeda makes no representations about whether investigational compounds or unapproved uses will be approved by the FDA.
Annual meeting dedicated to the advancement of neuromuscular, musculoskeletal, and electrodiagnostic medicine.

National conference of IgNS bringing together professionals and practitioners from all disciplines and clinical specialties to advance Ig therapy practice, while providing networking and comprehensive education opportunities.
Global meeting featuring thought leaders from around the world focused on advancing patient care in allergy and immunology.

Annual conference that invites state-of-the-art speakers from around the world to discuss basic science and clinical topics on allergy, asthma, and immunology.
The largest peripheral nerve meeting globally, offering the latest international research across specialties in peripheral neuropathy.

Regional annual conference discussing the most current information from top experts in allergy, asthma, and immunology.
Annual meeting with the goal of providing a stimulating forum with presentations and discussions on the latest advances in clinical immunology, including primary
immunodeficiencies and immune dysregulatory diseases.
Brings together home and alternate site infusion professionals for four days of networking, education, and exhibits. The expo features companies displaying the latest products and services supporting the industry.
Each year, the American Academy of Neurology's Annual Meeting offers a robust lineup of diverse learning opportunities covering nearly every topic and subspecialty, helping you stay up to date on the latest trusted science and essential education.
Semi-annual CIIC conferences provide a unique opportunity to network with experts and prospective partners in the allergy, asthma and immunology space.
Premier global educational event for allergists and immunologists, with thousands of attendees each year, discussing allergies, asthma, and immune deficiency disorders.

Global meeting featuring thought leaders from around the world focused on advancing patient care in allergy and immunology.
Annual meeting dedicated to the advancement of neuromuscular, musculoskeletal, and electrodiagnostic medicine.
Annual event with more than 2,500 members and non-members of the AMCP to engage on the latest innovations and most intentional networking in managed care pharmacy.

National conference of IgNS bringing together professionals and practitioners from all disciplines and clinical specialties to advance Ig therapy practice, while providing networking and comprehensive education opportunities.
Watch videos focused on Hereditary Angioedema (HAE).
Learn about Hereditary Angioedema (HAE), including the prevalence of the three types of HAE and their causes.
Learn about the delayed and misdiagnosis of Hereditary Angioedema (HAE) in patients presenting with abdominal symptoms.
Find materials to help foster a deeper understanding of Hereditary Angioedema (HAE).
An overview of HAE epidemiology, pathophysiology, signs and symptoms, diagnosis, and burden of disease.
An overview of HAE misdiagnosis, causes of angioedema, types of HAE, and diagnosing/testing for HAE.
An overview of HAE with nC1-INH including prevalence, pathophysiology, diagnosis, and subtypes.
An overview of HAE in pediatric patients including diagnosis, clinical presentation, and disease burden.
Click the button below to send us your Medical Inquiry. We are committed to responding to your inquiries, and will get back to you as soon as possible.
Contact us using the number below to:
Speak with a Medical Information Specialist Monday-Friday 8am-6pm ET.